Tutorial 7: Build and Visualize Hi-M Contact Matrices
Objective
Convert chromatin traces into pairwise distance matrices and contact probability matrices, then visualize them as publication-quality heatmaps.
This tutorial covers two scripts:
``trace_to_matrix`` — computes single-cell pairwise distance (PWD) matrices from a trace file and produces five plots (Hi-M contact matrix, PWD median, PWD KDE, N-matrix, distance histograms).
``plot_him_matrix`` — re-plots a Hi-M matrix from the saved
.npydata with custom options (colormap, color range, mode, triangular display, NaN threshold…).
Scientific context
Contact matrices summarize pairwise interactions across all traces:
Contact probability (Hi-M): fraction of traces where two barcodes are closer than a distance threshold (default 0.25 µm).
PWD median / KDE: ensemble pairwise distance estimated via median or kernel density peak.
N-matrix: number of valid (non-NaN) measurements per barcode pair — reveals data coverage.
These matrices reveal chromatin folding patterns such as topologically associating domains (TADs), long-range loops, and compartments.
Input
We use the Pdx1-positive trace file produced by Tutorial 6 (mask assignment + split):
File |
Description |
|---|---|
|
Traces inside the Pdx1 mask (single ROI) |
[1]:
import matplotlib.pyplot as plt
import matplotlib.image as mpimg
data_path = "/home/devos/Documents/data_to_compare_pdx1/PDX1"
# Pdx1-positive traces from Tutorial 6
input_trace = f"{data_path}/merged_traces_split.ecsv"
# Base name for all matrix outputs
matrix_base = f"{data_path}/merged_traces_split_Matrix"
print(f"Input trace: {input_trace}")
Input trace: /home/devos/Documents/data_to_compare_pdx1/PDX1/merged_traces_split.ecsv
Step 1: Build matrices with trace_to_matrix
trace_to_matrix reads a trace file and computes:
A 3D single-cell PWD matrix (shape: n_barcodes × n_barcodes × n_traces) saved as
.npyAn N-matrix (count of valid measurements per barcode pair) saved as
.npyA unique barcodes list saved as
.ecsvFive plots: Hi-M contact matrix, PWD median, PWD KDE, N-matrix, and distance histograms
Parameters
Option |
Default |
Description |
|---|---|---|
|
— |
Input trace file (ECSV) |
|
|
Maximum distance allowed (discard pairs beyond this) |
|
|
Output folder (for data files) |
[2]:
!trace_to_matrix --input {input_trace}
$ trace file to process= ['/home/devos/Documents/data_to_compare_pdx1/PDX1/merged_traces_split.ecsv']
$ Importing table from pyHiM format
Successfully loaded trace table: /home/devos/Documents/data_to_compare_pdx1/PDX1/merged_traces_split.ecsv
$ Found 23 barcodes and 3828 traces. INFO
> Processing traces... INFO
100%|██████████████████████████████████████| 3828/3828 [00:05<00:00, 685.76it/s]
100%|███████████████████████████████████████████| 23/23 [00:22<00:00, 1.03it/s]
/home/devos/Repo/traceratops/.venv/lib/python3.11/site-packages/numpy/lib/_nanfunctions_impl.py:1215: RuntimeWarning: All-NaN slice encountered
return fnb._ureduce(a, func=_nanmedian, keepdims=keepdims,
100%|███████████████████████████████████████████| 23/23 [02:24<00:00, 6.30s/it]
Output figure: /home/devos/Documents/data_to_compare_pdx1/PDX1/merged_traces_split_Matrix_PWDhistograms.png
$ saved: /home/devos/Documents/data_to_compare_pdx1/PDX1/merged_traces_split_Matrix_PWDscMatrix.npy
$ saved: /home/devos/Documents/data_to_compare_pdx1/PDX1/merged_traces_split_Matrix_uniqueBarcodes.ecsv
$ saved: /home/devos/Documents/data_to_compare_pdx1/PDX1/merged_traces_split_Matrix_Nmatrix.npy
Processed <1> trace(s)
Finished execution
Output files
All outputs use the pattern <input_basename>_Matrix_<suffix>:
File |
Format |
Description |
|---|---|---|
|
NumPy binary |
3D single-cell pairwise distance matrix |
|
NumPy binary |
2D N-matrix (data coverage) |
|
Text |
List of unique barcode IDs |
|
Image |
Contact probability heatmap |
|
Image |
PWD matrix (median) |
|
Image |
PWD matrix (KDE peak) |
|
Image |
N-matrix heatmap |
|
Image |
Distance histograms for all barcode pairs |
Step 2: Hi-M contact probability matrix
The Hi-M matrix shows the fraction of traces where each barcode pair is closer than a distance threshold (default: 0.25 µm). Higher values (warm colors) indicate more frequent contacts.
[3]:
img = mpimg.imread(f"{matrix_base}_HiMmatrix.png")
fig, ax = plt.subplots(figsize=(10, 8))
ax.imshow(img)
ax.axis('off')
ax.set_title("Hi-M Contact Probability Matrix", fontsize=14)
plt.tight_layout()
plt.show()
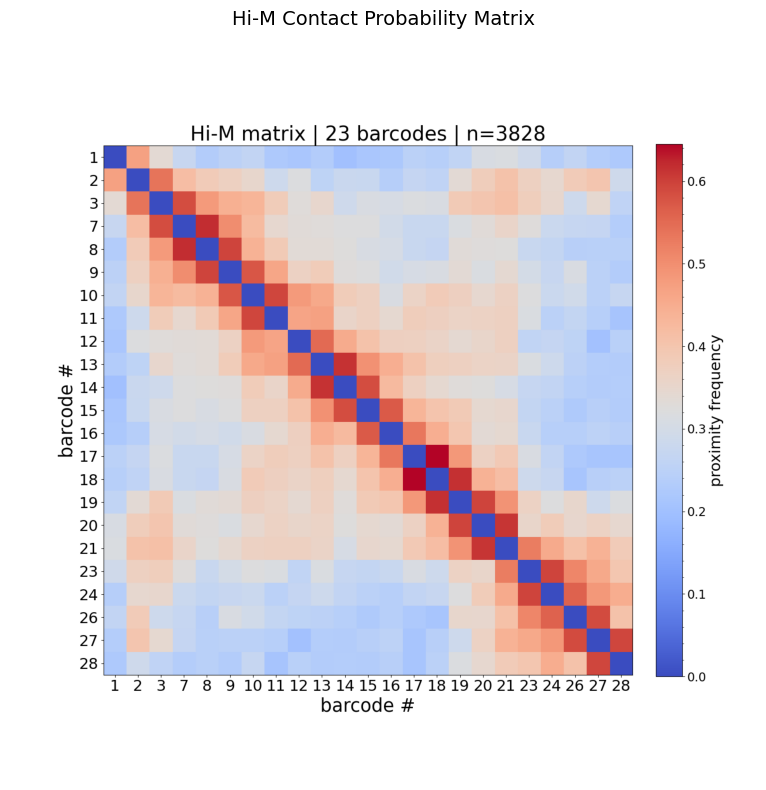
How to read this matrix:
Axes show barcode IDs (genomic loci along the chromatin fiber).
The diagonal is always high (a barcode is always close to itself).
Off-diagonal warm spots indicate frequent spatial proximity between distant genomic loci → potential loops or TAD boundaries.
Blocks along the diagonal suggest topologically associating domains (TADs).
Step 3: Pairwise distance matrices
Two PWD representations are computed:
Median: robust central tendency of 3D distances per barcode pair.
KDE: peak of the kernel density estimate — captures the most probable distance, useful when distributions are skewed.
[4]:
fig, axes = plt.subplots(1, 2, figsize=(20, 8))
for ax, suffix, title in zip(
axes,
["_PWDmatrixMedian.png", "_PWDmatrixKDE.png"],
["PWD Matrix (Median)", "PWD Matrix (KDE)"]
):
img = mpimg.imread(f"{matrix_base}{suffix}")
ax.imshow(img)
ax.axis('off')
ax.set_title(title, fontsize=14)
plt.tight_layout()
plt.show()
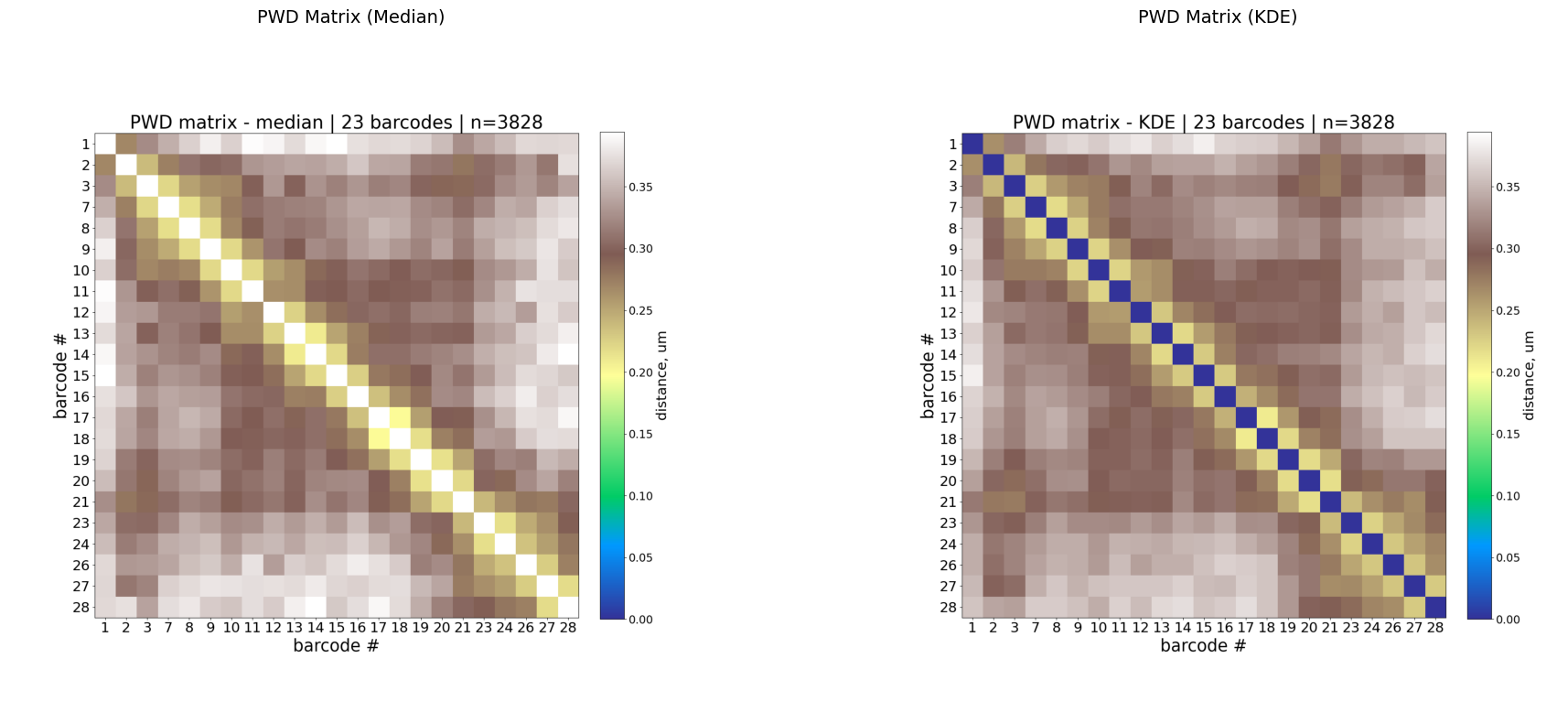
Interpretation:
Low values along the diagonal (warm colors in
terraincolormap) = nearby genomic loci are physically close (expected).Off-diagonal low-distance spots indicate long-range contacts.
KDE is often less noisy than median for sparse data.
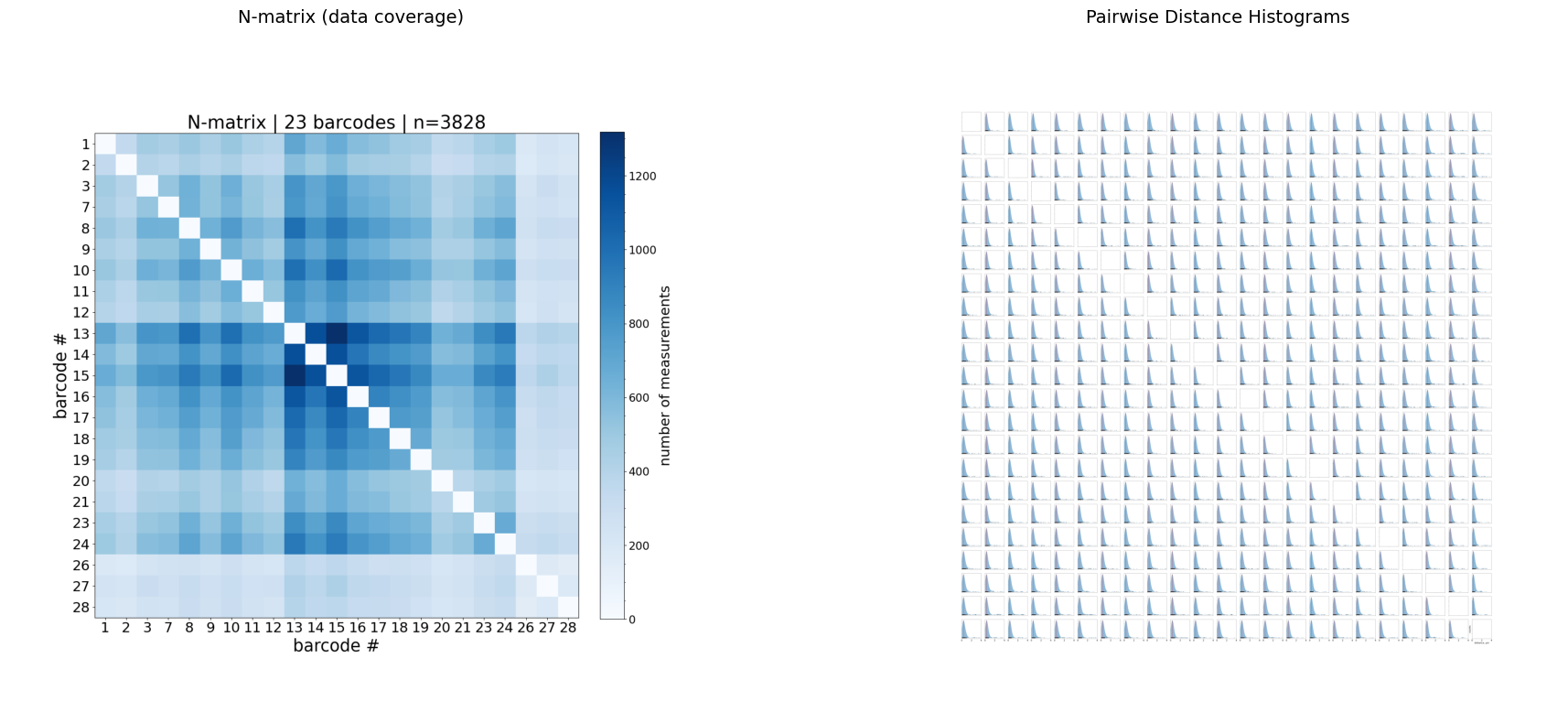
Step 4: N-matrix and distance histograms
The N-matrix shows how many valid (non-NaN) pairwise measurements exist for each barcode pair. Low N values indicate poor coverage — those entries should be interpreted with caution.
The distance histograms show the full distribution (KDE) of 3D distances for every barcode pair.
[5]:
fig, axes = plt.subplots(1, 2, figsize=(20, 8))
for ax, suffix, title in zip(
axes,
["_Nmatrix.png", "_PWDhistograms.png"],
["N-matrix (data coverage)", "Pairwise Distance Histograms"]
):
img = mpimg.imread(f"{matrix_base}{suffix}")
ax.imshow(img)
ax.axis('off')
ax.set_title(title, fontsize=14)
plt.tight_layout()
plt.show()
Step 5: Re-plot with plot_him_matrix
plot_him_matrix lets you re-visualize a previously computed matrix with custom display options, without re-running the full computation.
It reads:
The 3D single-cell PWD matrix (
*_PWDscMatrix.npy)The unique barcodes list (
*_uniqueBarcodes.ecsv)
Key parameters
Option |
Default |
Description |
|---|---|---|
|
— |
Single-cell PWD matrix ( |
|
— |
Unique barcodes file |
|
|
|
|
|
Contact distance threshold in µm (for |
|
auto |
Colormap range |
|
|
Matplotlib colormap name |
|
|
Mask bins where NaN percentage exceeds this value (0–1) |
|
off |
Show only upper or lower triangle |
|
|
|
|
|
Output folder for the plot |
|
|
|
Example: proximity matrix with custom threshold
Recompute the contact probability using a different distance threshold (e.g. 0.30 µm instead of the default 0.25 µm):
[6]:
sc_matrix = f"{matrix_base}_PWDscMatrix.npy"
barcodes = f"{matrix_base}_uniqueBarcodes.ecsv"
!plot_him_matrix -M {sc_matrix} -B {barcodes} --mode proximity -T 0.30 -O {data_path}/plots
Output path: /home/devos/Documents/data_to_compare_pdx1/PDX1/plots
$ Matrix loaded: /home/devos/Documents/data_to_compare_pdx1/PDX1/merged_traces_split_Matrix_PWDscMatrix.npy
$ Unique barcodes loaded: [1, 2, 3, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 23, 24, 26, 27, 28]
$ averaging method: proximity
$ calculating contact probability matrix
Output data: /home/devos/Documents/data_to_compare_pdx1/PDX1/plots/Fig_merged_traces_split_Matrix_PWDscMatrix_proximity_norm_T0.3_0.31-0.75.npy
[7]:
import glob
plots_path = f"{data_path}/plots"
matches = sorted(glob.glob(f"{plots_path}/Fig_*_proximity_norm_T0.3_*.png"))
if matches:
img = mpimg.imread(matches[0])
fig, ax = plt.subplots(figsize=(10, 8))
ax.imshow(img)
ax.axis('off')
ax.set_title("Proximity matrix (T = 0.30 µm)", fontsize=14)
plt.tight_layout()
plt.show()
print(f"Plot: {matches[0]}")
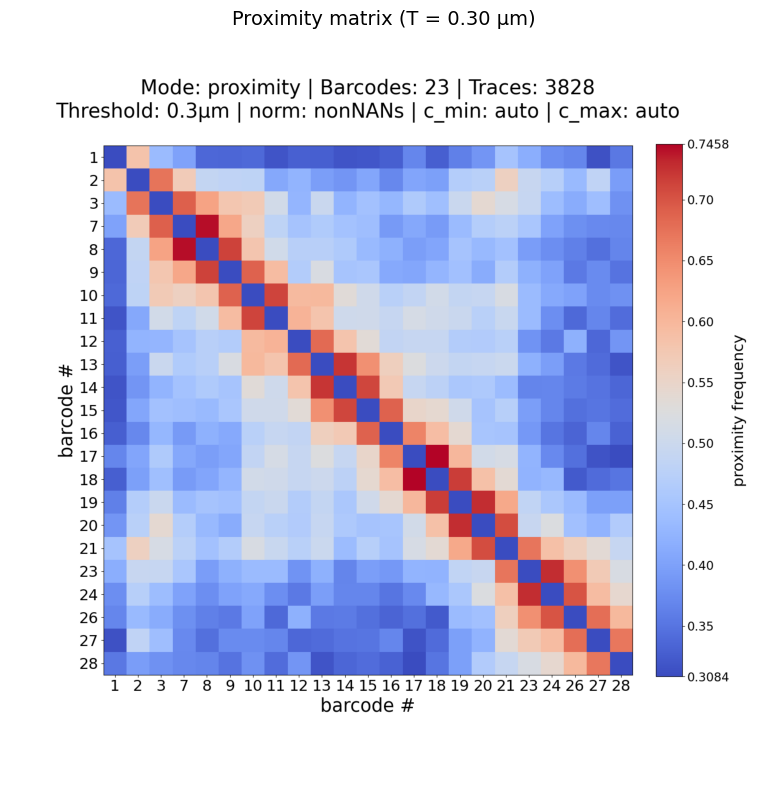
Plot: /home/devos/Documents/data_to_compare_pdx1/PDX1/plots/Fig_merged_traces_split_Matrix_PWDscMatrix_proximity_norm_T0.3_0.31-0.75.png
Example: median PWD matrix
[8]:
!plot_him_matrix -M {sc_matrix} -B {barcodes} --mode median -O {data_path}/plots
Output path: /home/devos/Documents/data_to_compare_pdx1/PDX1/plots
$ Matrix loaded: /home/devos/Documents/data_to_compare_pdx1/PDX1/merged_traces_split_Matrix_PWDscMatrix.npy
$ Unique barcodes loaded: [1, 2, 3, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 23, 24, 26, 27, 28]
$ averaging method: median
/home/devos/Repo/traceratops/.venv/lib/python3.11/site-packages/numpy/lib/_nanfunctions_impl.py:1215: RuntimeWarning: All-NaN slice encountered
return fnb._ureduce(a, func=_nanmedian, keepdims=keepdims,
Output data: /home/devos/Documents/data_to_compare_pdx1/PDX1/plots/Fig_merged_traces_split_Matrix_PWDscMatrix_median_norm_0.20-0.40.npy
[9]:
matches = sorted(glob.glob(f"{plots_path}/Fig_*_median_norm_*.png"))
if matches:
img = mpimg.imread(matches[0])
fig, ax = plt.subplots(figsize=(10, 8))
ax.imshow(img)
ax.axis('off')
ax.set_title("PWD Median matrix", fontsize=14)
plt.tight_layout()
plt.show()
print(f"Plot: {matches[0]}")
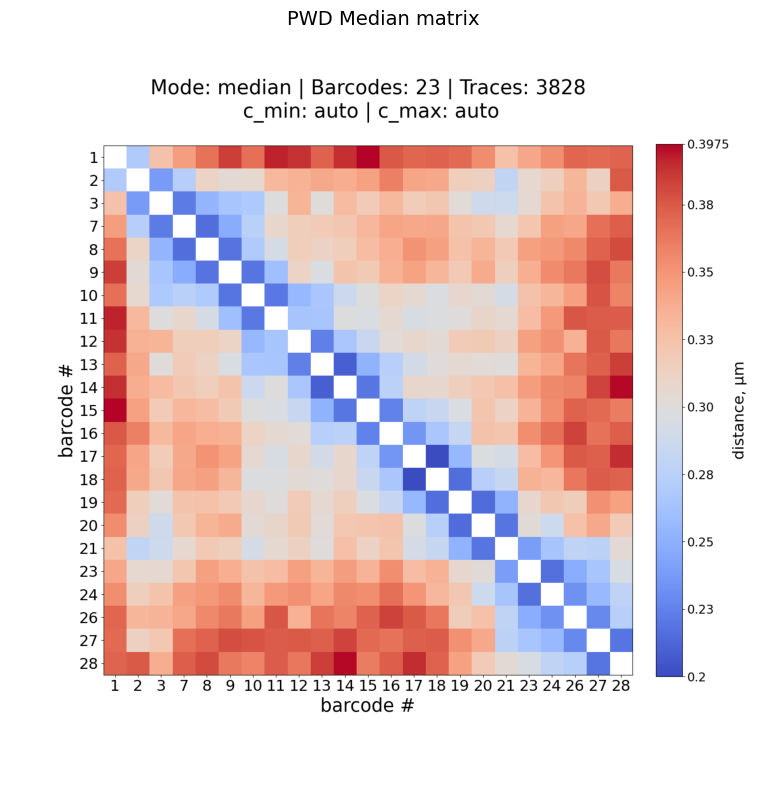
Plot: /home/devos/Documents/data_to_compare_pdx1/PDX1/plots/Fig_merged_traces_split_Matrix_PWDscMatrix_median_norm_0.20-0.40.png
Summary
Workflow
trace file (.ecsv)
│
▼
trace_to_matrix → 5 plots + 3 data files
│
▼
plot_him_matrix (optional) → re-plot with custom options
Produced by trace_to_matrix
Plot |
What it shows |
|---|---|
|
Contact probability (proximity frequency) |
|
Pairwise distance — median |
|
Pairwise distance — KDE peak |
|
Data coverage (N measurements per pair) |
|
Full distance distributions (all pairs) |
Customization with plot_him_matrix
Use case |
Command |
|---|---|
Change distance threshold |
|
Median distance matrix |
|
KDE distance matrix |
|
Custom colormap |
|
Mask sparse bins |
|
Triangular plot |
|
PDF for publication |
|