Tutorial 8: Colocalization Analysis (2-way & 3-way)
Objective
Beyond pairwise distance matrices (Tutorial 7), chromatin tracing data allows us to ask: given a specific genomic locus (anchor), which other loci are physically close to it?
This tutorial covers two complementary analyses:
2-way colocalization (
plot_4m) — For a given anchor barcode, compute the frequency at which each other barcode is within a distance cutoff. Produces a 1D profile (4M plot).3-way colocalization (
trace_3way_coloc) — For a given anchor barcode, compute the frequency at which pairs of other barcodes are both within the distance cutoff simultaneously. Produces a 2D heatmap.
Both scripts include bootstrapping to estimate statistical confidence (mean ± SEM).
Scientific context
2-way (4M): reveals which loci interact with a chosen viewpoint — analogous to 4C/Capture-C.
3-way: identifies higher-order contacts where 3 loci converge simultaneously — evidence for transcription factories, chromatin hubs, or phase-separated condensates.
Input
We use the Pdx1-positive trace file from Tutorial 6:
File |
Description |
|---|---|
|
Traces inside the Pdx1 mask |
[1]:
import matplotlib.pyplot as plt
import matplotlib.image as mpimg
import glob
data_path = "/home/devos/Documents/data_to_compare_pdx1/PDX1"
input_trace = f"{data_path}/merged_traces_split.ecsv"
print(f"Input trace: {input_trace}")
Input trace: /home/devos/Documents/data_to_compare_pdx1/PDX1/merged_traces_split.ecsv
Step 1: 2-way colocalization with plot_4m
plot_4m computes, for a chosen anchor barcode, how frequently every other barcode is found within a distance cutoff. This is repeated over bootstrap iterations to obtain mean ± SEM.
The result is a 4M plot: a 1D profile where the X-axis is the barcode number and the Y-axis is the colocalization frequency with the anchor.
Parameters
Option |
Default |
Description |
|---|---|---|
|
— |
Input trace file (ECSV) |
|
— |
One or more anchor barcode numbers (space-separated) |
|
|
Distance threshold in µm |
|
|
Number of bootstrap iterations |
|
|
Base name for output plots |
|
auto |
X-axis range for the plot |
[2]:
# 2-way colocalization: anchor at barcode 10, distance cutoff 0.25 µm
!plot_4m --input {input_trace} --anchors 10 --cutoff 0.25 --bootstrapping_cycles 50 --output {data_path}/coloc_4m.png
$ Processing trace file: /home/devos/Documents/data_to_compare_pdx1/PDX1/merged_traces_split.ecsv
$ Importing table from pyHiM format
Successfully loaded trace table: /home/devos/Documents/data_to_compare_pdx1/PDX1/merged_traces_split.ecsv
$ Processing anchors: [10]
The output is saved as coloc_4m_anchor_10.png (the anchor ID is appended automatically).
[3]:
plot_file = f"{data_path}/coloc_4m_anchor_10.png"
img = mpimg.imread(plot_file)
fig, ax = plt.subplots(figsize=(12, 5))
ax.imshow(img)
ax.axis('off')
ax.set_title("4M plot: colocalization with anchor barcode 10", fontsize=14)
plt.tight_layout()
plt.show()
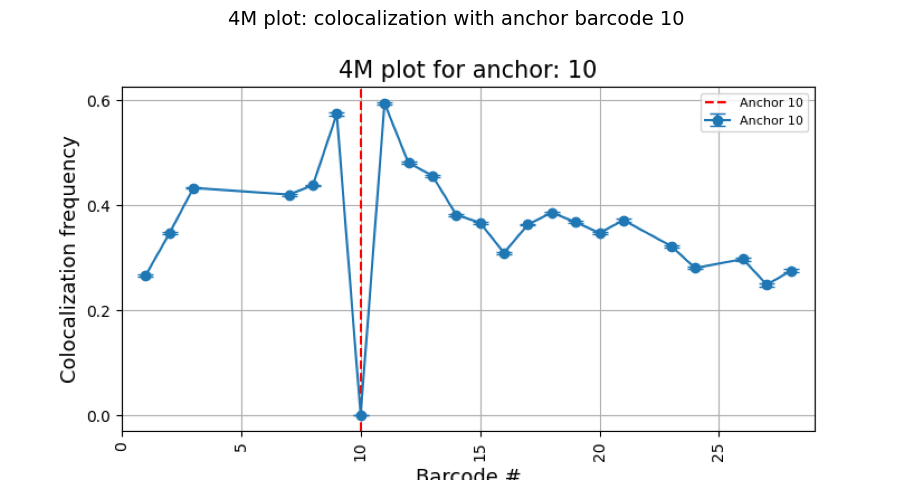
How to read the 4M plot:
Each point is a barcode; the Y-value is the fraction of traces where that barcode is within 0.25 µm of the anchor.
Error bars show the bootstrap SEM.
The red dashed line marks the anchor barcode itself.
Barcodes near the anchor on the genome (close barcode numbers) are expected to have higher frequencies.
A barcode far on the genome but with high frequency suggests a long-range contact.
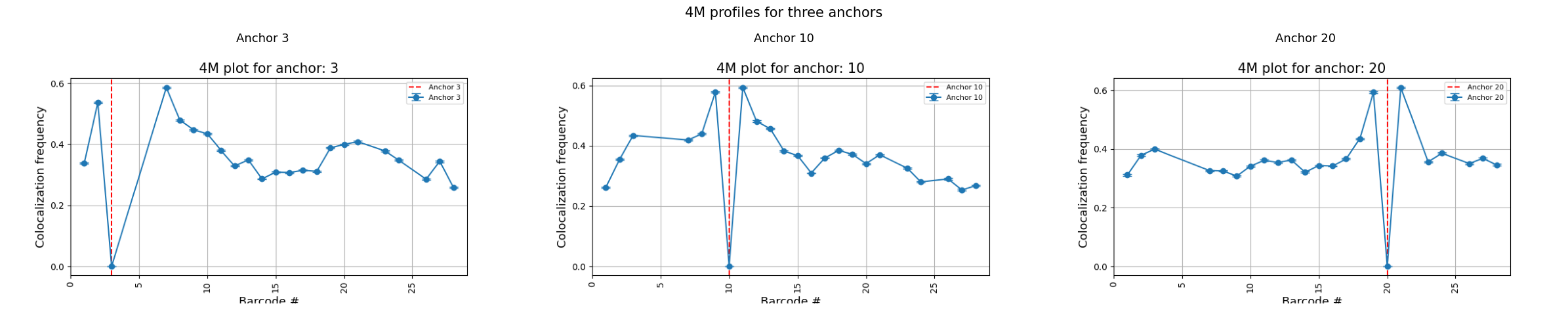
Multiple anchors
You can compute 4M profiles for several anchors in a single run. Each anchor produces its own plot.
[4]:
# Multiple anchors in one run
!plot_4m --input {input_trace} --anchors 3 10 20 --cutoff 0.25 --bootstrapping_cycles 50 --output {data_path}/coloc_4m_multi.png
$ Processing trace file: /home/devos/Documents/data_to_compare_pdx1/PDX1/merged_traces_split.ecsv
$ Importing table from pyHiM format
Successfully loaded trace table: /home/devos/Documents/data_to_compare_pdx1/PDX1/merged_traces_split.ecsv
$ Processing anchors: [3, 10, 20]
[5]:
# Display all anchor plots side by side
anchors = [3, 10, 20]
fig, axes = plt.subplots(1, 3, figsize=(24, 5))
for ax, anchor in zip(axes, anchors):
plot_file = f"{data_path}/coloc_4m_multi_anchor_{anchor}.png"
img = mpimg.imread(plot_file)
ax.imshow(img)
ax.axis('off')
ax.set_title(f"Anchor {anchor}", fontsize=13)
fig.suptitle("4M profiles for three anchors", fontsize=15)
plt.tight_layout()
plt.show()
Step 2: 3-way colocalization with trace_3way_coloc
trace_3way_coloc extends the analysis to three-body contacts. Given an anchor barcode, it checks every pair of other barcodes (i, j) and asks: in what fraction of traces are anchor, i, and j all within the distance cutoff simultaneously?
The result is a symmetric heatmap where each cell (i, j) shows the 3-way colocalization frequency. A second heatmap shows the bootstrap SEM.
Parameters
Option |
Default |
Description |
|---|---|---|
|
— |
Input trace file (ECSV) |
|
— |
One or more anchor barcode numbers |
|
|
Distance threshold in µm |
|
|
Number of bootstrap iterations |
|
|
Base name for output files |
|
auto |
Colormap range |
[6]:
# 3-way colocalization: anchor at barcode 10
!trace_3way_coloc --input {input_trace} --anchors 10 --cutoff 0.25 --bootstrapping_cycles 50 --output {data_path}/coloc_3way.png
>> Processing file: /home/devos/Documents/data_to_compare_pdx1/PDX1/merged_traces_split.ecsv
$ Importing table from pyHiM format
Successfully loaded trace table: /home/devos/Documents/data_to_compare_pdx1/PDX1/merged_traces_split.ecsv
Using distance cutoff: 0.25 µm
Performing 50 bootstrap iterations
Running analysis for anchor: 10
Saved three-way co-localization heatmap to: /home/devos/Documents/data_to_compare_pdx1/PDX1/coloc_3way_merged_traces_split_anchor_10
Saved SEM heatmap to: /home/devos/Documents/data_to_compare_pdx1/PDX1/coloc_3way_merged_traces_split_anchor_10_sem.png
This produces:
Output file |
Description |
|---|---|
|
3-way frequency heatmap |
|
Frequency matrix (NumPy) |
|
Bootstrap SEM heatmap |
[7]:
# Find and display the 3-way heatmap (frequency and SEM)
freq_matches = sorted(glob.glob(f"{data_path}/coloc_3way_*_anchor_10.png"))
sem_matches = sorted(glob.glob(f"{data_path}/coloc_3way_*_anchor_10_sem.png"))
if freq_matches and sem_matches:
fig, axes = plt.subplots(1, 2, figsize=(20, 8))
img_freq = mpimg.imread(freq_matches[0])
axes[0].imshow(img_freq)
axes[0].axis('off')
axes[0].set_title("3-way colocalization frequency", fontsize=14)
img_sem = mpimg.imread(sem_matches[0])
axes[1].imshow(img_sem)
axes[1].axis('off')
axes[1].set_title("Bootstrap SEM", fontsize=14)
plt.tight_layout()
plt.show()
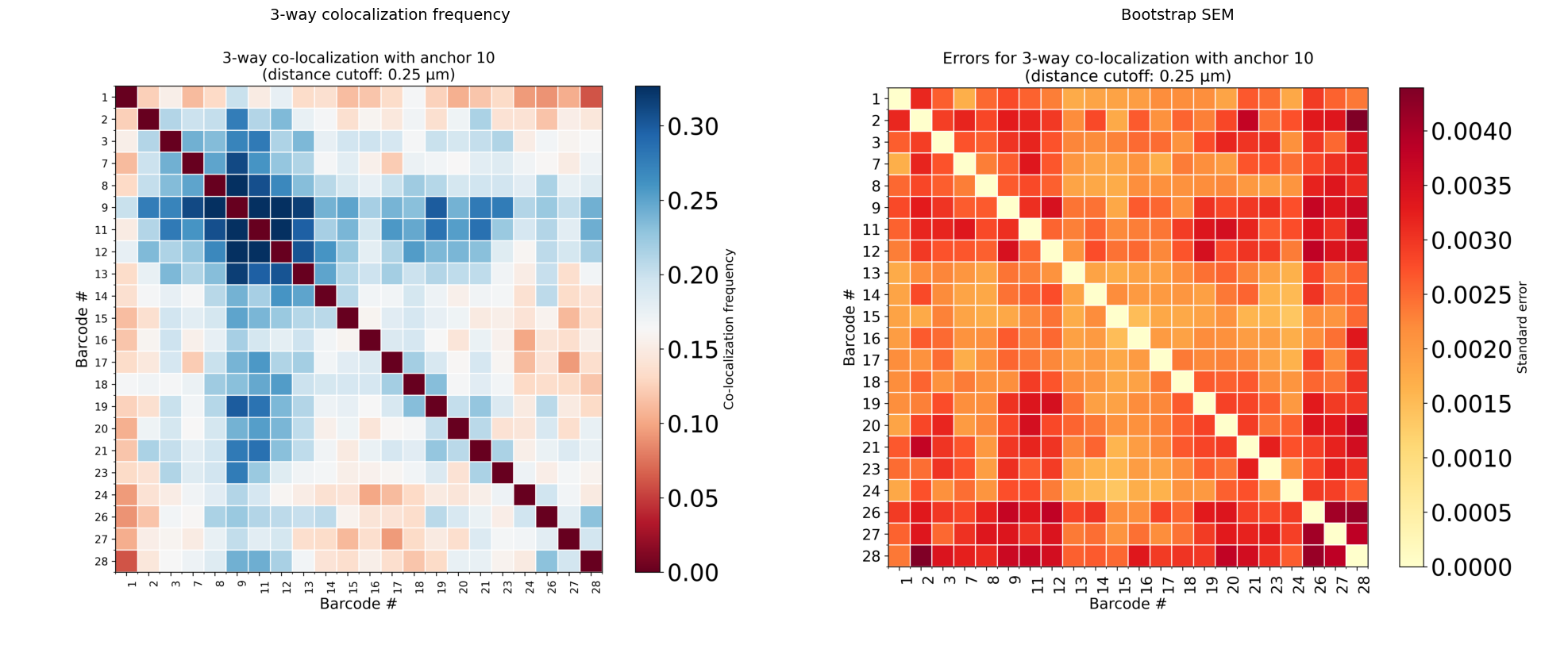
How to read the 3-way heatmap:
Axes show barcode IDs (excluding the anchor).
Cell (i, j) = fraction of traces where anchor, barcode i, and barcode j are all three within the distance cutoff.
Black crosshair marks the anchor barcode position.
High values off the diagonal reveal three-body hubs where three distant loci converge.
The SEM heatmap helps identify which entries are statistically robust (low SEM).
Step 3: Varying the distance cutoff
The distance cutoff has a strong effect on colocalization frequencies. A small cutoff (e.g. 0.15 µm) captures only very tight contacts, while a larger one (e.g. 0.35 µm) includes more diffuse proximity. Try different values to see how the patterns change:
# Tight cutoff
plot_4m --input Trace.ecsv --anchors 10 --cutoff 0.15 --output coloc_tight.png
# Relaxed cutoff
plot_4m --input Trace.ecsv --anchors 10 --cutoff 0.35 --output coloc_relaxed.png
A robust interaction should be visible across a range of cutoffs.
Summary
Workflow
trace file (.ecsv)
│
├─── plot_4m → 1D profile (anchor vs each barcode)
│
└─── trace_3way_coloc → 2D heatmap (anchor vs pairs) + SEM
Commands reference
# 2-way (4M profile)
plot_4m --input Trace.ecsv --anchors 10 --cutoff 0.25 --bootstrapping_cycles 50 --output 4m.png
# 3-way (heatmap)
trace_3way_coloc --input Trace.ecsv --anchors 10 --cutoff 0.25 --bootstrapping_cycles 50 --output 3way.png
Key differences
|
|
|
|---|---|---|
Question |
Is barcode X close to anchor? |
Are barcodes X and Y both close to anchor? |
Output |
1D line plot |
2D symmetric heatmap |
Complexity |
O(n) per trace |
O(n²) per trace |
Detects |
Pairwise interactions |
Higher-order hubs |
Notes
Bootstrap cycles: 10 is fast for exploration; use 100–1000 for publication.
Multiple anchors: both scripts accept
--anchors 3 10 20to process several viewpoints in one run.Large datasets: 3-way analysis is computationally heavier than 2-way. For many barcodes, consider running on a filtered/split subset.